The b -globin gene family and Sickle Cell Anemia
The human genome consists of approximately 3 billion base pairs. Until recently, scientists had predicted that this large amount of DNA would encode almost 100,000 different genes; however, now that the sequencing of the entire human genome is almost complete, that number has dropped to only 30,000 genes. In fact, close analysis of the genome has shown that well over 90 percent of the genome consists of non-functional DNA.
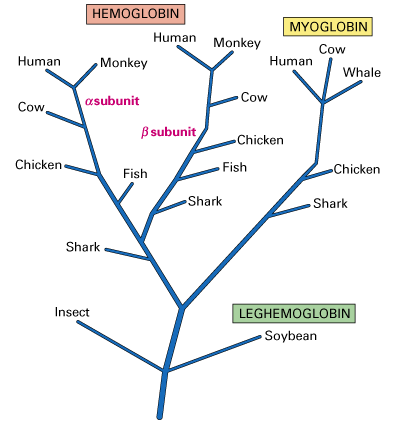
Of the remaining functional DNA, 25-50 percent of the genes encoding proteins are found only once in the haploid genome and are known as solitary genes. Frequently, the DNA surrounding a particular gene contains sequences that are close, but not identical, copies of the gene. These multiple copies are believed to be the result of duplication and divergence. This is the process by which a single gene is first duplicated and then undergoes selective pressure to mutate into a gene that is similar, but not identical, in sequence to its ancestral gene. An evolutionary tree predicting the development of the globin family of genes is shown below and illustrates the divergence of genes from leghemoglobin in plants to hemoglobin and myoglobin.
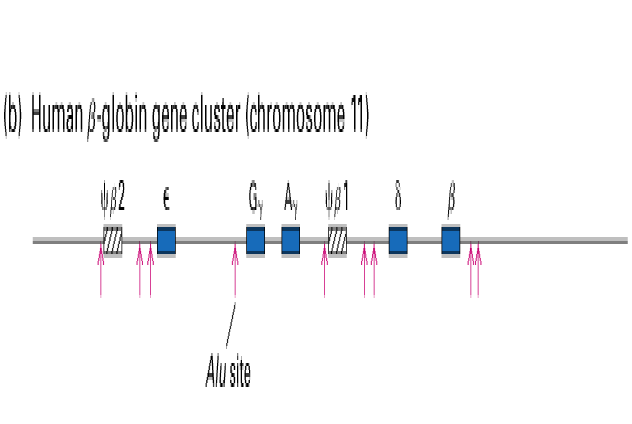
A set of duplicated genes that encode proteins with similar but nonidentical amino acid sequences is known as a gene family (Lodish et.al., 2000). One example commonly used to describe a gene family is the human b -globin gene cluster found on chromosome 11. As shown in the figure below, the globin family consists of five functional genes (blue boxes) and two pseudogenes (diagonal lines).
All of the hemoglobins encoded by these different genes function to carry oxygen in the blood; however, each gene exhibits specific variations in function. For example, the Ag and Gg genes are expressed only during fetal development. These hemoglobin proteins have a higher binding affinity for oxygen than the adult hemoglobins encoded by the b and d genes. This increased binding allows the fetus to successfully extract oxygen from the blood without competing with the mother. Adult hemoglobin has a lower oxygen affinity allowing better release of oxygen to tissue, especially muscle.
Two regions of the globin gene family contain nonfunctional sequences known as pseudogenes. These genes are similar to their functional globin gene counterparts but are no longer transcribed into mRNA because of changes in sequence that have occurred during the course of their evolution.
To form a functional hemoglobin molecule, two b -globin polypeptides combine with two a -globin polypeptides (encoded by another gene family) and four small heme groups. Even a slight change in one of the genes encoding a subunit of the hemoglobin molecule can have disastrous results. Sickle cell anemia, for example, occurs in 1 in 500 individuals of African descent and characterized by red blood cells that are rigid and sickled in shape.
The difficulty these sickle cells have in moving through the circulatory system has numerous effects on those who suffer from the disease.
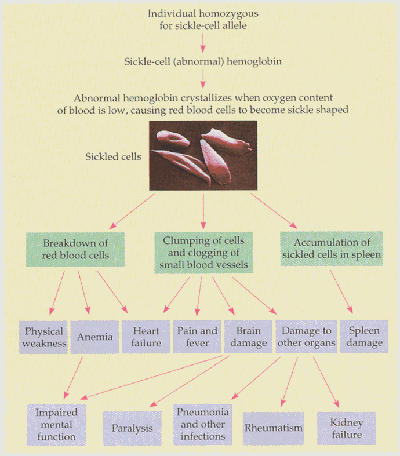
Believe or not, all of the symptoms listed in the figure above are caused by a single point mutation in codon six of the b -globin gene from glutamic acid to valine (See figure below). This small difference produces a mutant protein that is insoluble in the red blood cell, and consequently, forms a crystalline structure.
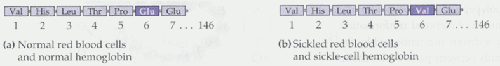
There are, however, advantages to carrying only one copy of the mutant b -globin gene. These heterozygote individuals do not suffer from the disease and exhibit increased resistance to malaria because the parasite which causes the disease is unable to reproduce in the red blood cells of heterozygote individuals. Malaria occurs most frequently in tropical regions near the equator which helps to explain why those of African descent are more commonly affected.
Considering the devastating effects of sickle cell anemia, parents may want to determine if they are carriers of the disease before they decide to have children. If two carriers mate, 25 percent of their children will be homozygous for the mutant gene. The use of restriction enzymes now enables carriers to be determined by examining RFLPs, or restriction fragment length polymorphisms. Mutations in DNA that add or delete a restrictions site can be determined by analysis using gel electrophoresis after digesting DNA with a restriction enzyme. First compare the restriction sites (shown in blue) found on the wild-type and mutant b -globin genes:
Notice that the point mutation in codon six eliminates one of the restriction sites normally found in the wild-type copy of the b -globin gene. If these two genes were digested with restriction enzyme and run out on a gel you would expect the following results:
The b -globin gene family and b -thalessemia
Sickle cell anemia is not the only disease caused by a mutation in the b -globin gene. A second disease known as beta-thalessemia occurs as a result of several mutations in the b -globin gene and ultimately leads to abrogated production of the b -globin protein. There are over 500 b -thalessemia causing mutations that have been identified. These mutations do not affect the structure of hemoglobin so sickled blood cells are not present; instead, suffers of b -thalessemia lack functional hemoglobin.
The symptoms of this disease vary greatly depending on the amount of functional hemoglobin that is produced. People with mild cases of b -thalessemia often display no symptoms and are never diagnosed with the disease. On the other hand, those with severe cases require constant blood tranfusions beginning at age one to two months to supply them with functional hemoglobin required to carry oxygen throughout the body.
